2. Norma geral IEC 60601-1
Apresentação
Neste capítulo será apresentada a norma geral da série IEC 60601. Essa norma reúne os ensaios obrigatórios a serem realizados em todo e qualquer equipamento médico para fins de avaliar sua segurança básica e seu desempenho essencial. A versão atual incorpora o conceito de gerenciamento de risco e ciclo de vida, assim como novidades significativas em relação às versões anteriores da norma.
2.1. Histórico
Em 1976, o subcomitê técnico da IEC STC62A publicou a primeira edição do relatório técnico (Technical Report) IEC/TR 60513 – Basic aspects of the safety philosophy for electrical equipment used in medical practice (“Aspectos básicos da filosofia de segurança de equipamentos elétricos utilizados na prática médica”). Esse documento foi a gênese da norma técnica IEC 60601-1, dentre outros documentos da série IEC 60601. A IEC 60601-1 foi publicada inicialmente em 1977 (edição 1.0) e se tornou, desde então, a mais completa referência mundial em se tratando de equipamentos eletromédicos. A edição 1.1 (primeira revisão da edição 1.0) foi publicada em 1984. Já a edição 2.0 foi publicada em 1988 e revisada em 1991 (2.1) e 1995 (2.2). A terceira edição foi publicada em 2005 (3.0).
A versão atual dessa norma é a IEC 60601-1:2005 +A1:2012 – Medical electrical equipment – Part 1: General requirements for basic safety and essential performance. Ela corresponde à terceira edição revisada em 2012 (utilizando a nomenclatura da IEC, esta é a Edição 3.1). Como dado curioso, pouco antes da publicação da emenda à terceira edição foram apresentados 11.910 votos (comentários técnicos e editoriais) por parte dos diversos comitês nacionais que participaram dessa revisão, incluindo o Brasil. Ou seja, essa emenda foi muito bem analisada e comentada por especialistas do mundo todo.
Como já vimos no Capítulo 1, complementam a série 60601 as oito normas colaterais atualmente vigentes e as mais de sessenta normas particulares, cada qual responsável por detalhar os ensaios para os diversos tipos de equipamentos eletromédicos. Mas, além das normas vigentes, circunstancialmente as normas podem estar em vigência por um período posterior à publicação de uma edição mais recente. Isso acontece porque, em muitos casos, essas normas são utilizadas para certificação de equipamentos eletromédicos, e nem sempre a revisão das normas colaterais ou particulares acompanha o ritmo da revisão da norma geral ou vice-versa. Assim sendo, é possível haver uma norma particular que tenha sido revisada e publicada utilizando conceitos e requisitos previstos em uma norma colateral que ainda não foi publicada, mas está em revisão. Ademais, quando há uma quebra significativa de paradigmas em uma nova edição de uma norma técnica, há que se dar um tempo para que a indústria possa se adequar, portanto a edição antiga de determinada norma pode continuar válida para manter a conformidade. A IEC tem um termo específico para esse tipo de situação: DOCOPOCOSS (Date Of Cessation Of Presumption Of Conformity Of Superseded Standard, ou seja, data de cessação da presunção de conformidade de norma substituída). Isto é, uma norma que foi substituída por outra edição mais recente pode continuar sendo válida, por tempo determinado, para fins de avaliação da conformidade. No caso da série IEC 60601, o conjunto de normas válidas, incluindo emendas e normas em DOCOPOCOSS, supera 80 documentos.
2.2. Escopo
O escopo da terceira edição da IEC 60601-1 passou por significativas alterações em relação às edições anteriores. Originalmente, a norma se aplicava a equipamentos “sob supervisão médica”, ou seja, equipamentos que somente podem ser operados por profissional da área da saúde, principalmente médicos. Entretanto, percebeu-se que muitos equipamentos podem afetar significativamente a saúde de seus usuários se não forem certificados segundo os preceitos do desempenho essencial e segurança básica, mesmo não sendo necessário o monitoramento profissional contínuo e ininterrupto. Como exemplo, os termômetros clínicos (80601-2-56) não são exclusivamente utilizados sob supervisão médica, mas podem ser comercializados para qualquer cidadão e podem ter uso doméstico sem desvirtuar sua funcionalidade precípua. Alguns equipamentos podem ficar em operação sem supervisão ininterrupta, embora devam ser supervisionados por profissionais da saúde. Equipamentos como desfibriladores automáticos de emergência (60601-2-4), esfigmomanômetros (80601-2-30) e diversos equipamentos médicos para uso domiciliar estão nesse caso. Por essa razão, o escopo da série 60601 foi estendido para “equipamentos médicos elétricos” (medical electrical equipment), ou, como se usa corriqueiramente no Brasil, “equipamentos eletromédicos”.
Outro ponto aperfeiçoado no escopo da terceira edição da IEC 60601-1 foi a inclusão de “compensação ou alívio de doenças, lesões ou deficiências físicas” (compensation or alleviation of disease, injury or disability) na definição de equipamentos eletromédicos. Portanto, o escopo foi estendido para outras funcionalidades de equipamentos médicos, e não apenas a de tratar ou diagnosticar doenças. Por exemplo, aquecedores infantis (60601-2-21), cobertores e mantas (80601-2-35) ou camas hospitalares (60601-2-38) não são especificamente empregados no diagnóstico ou tratamento de pacientes, mas atuam de maneira coadjuvante no atendimento médico-hospitalar e na saúde como um todo. Após a revisão do escopo nessa terceira edição, esses equipamentos médicos puderam passar a ser incorporados à série IEC 60601.
A relação entre a norma geral e as normas colaterais também foi tratada na terceira edição. A partir de então, assim que uma norma colateral for publicada ela passa a ser, automaticamente, uma parte normativa integrante da norma geral. Assim sendo, muitas normas particulares passaram a ser revisadas para que as cláusulas da norma geral e as das normas colaterais fossem entendidas como um único documento normativo. Essa forma de entender e tratar as normas colaterais não estava muito clara até a publicação da terceira edição da norma geral.
Dentre todas as alterações significativas da terceira edição da IEC 60601-1, a que possivelmente gerou maior impacto na indústria foi a necessidade da introdução de gerenciamento de risco formal por parte dos fabricantes. Os aspectos do gerenciamento de risco serão apresentados mais à frente neste capítulo.
O escopo e o objetivo da terceira edição revisada da IEC 60601-1, bem como sua relação com as normas colaterais e particulares, estão explicados a seguir e foram traduzidos da versão em inglês (tradução do autor):
✓Escopo. Esta Norma Internacional é aplicável à SEGURANÇA BÁSICA e ao DESEMPENHO ESSENCIAL de EQUIPAMENTOS ELETROMÉDICOS e SISTEMAS ELETROMÉDICOS. Nos casos em que uma cláusula ou subcláusula seja aplicável apenas a equipamentos eletromédicos ou apenas a sistemas eletromédicos, o título e o conteúdo da cláusula ou subcláusula farão menção a isso. Se não for feita essa menção, o requisito será aplicado tanto para equipamentos quanto para sistemas eletromédicos. Perigos inerentes às funções fisiológicas de equipamentos ou sistemas eletromédicos não estão cobertos por requisitos específicos nesta norma, exceto os que constam nas cláusulas 7.2.13 (efeitos fisiológicos – avisos de segurança e alertas) e 8.4.1 (conexões ao paciente que transmitem corrente).
✓Objetivo. O objetivo desta norma é especificar requisitos gerais e serve como base para a aplicação das normas particulares.
✓Normas colaterais. Na série IEC 60601, normas colaterais especificam requisitos para segurança básica e desempenho essencial aplicáveis a subgrupos de equipamentos eletromédicos (equipamentos radiológicos, por exemplo) ou para características específicas que não sejam completamente abordadas na norma geral. Normas colaterais passam a ser consideradas requisitos normativos na data de sua publicação e devem ser aplicadas em conjunto com esta norma geral. Se uma norma colateral é aplicável a um equipamento ou sistema eletromédico para o qual existe uma norma particular, a norma particular tem prioridade sobre a norma colateral.
✓Normas particulares. Na série de normas 60601, normas particulares podem modificar, substituir ou eliminar requisitos desta norma geral conforme seja apropriado para o equipamento eletromédico em questão, além de poder acrescentar outros requisitos de segurança básica e desempenho essencial. Um requisito de uma norma particular tem prioridade sobre o respectivo requisito da norma geral.
2.3. Requisitos gerais
Equipamentos médicos diferem dos demais tipos de equipamentos ou produtos, por mais tecnológicos ou simples que possam ser. Em condições normais de uso pode-se, com alguma tranquilidade, separar os aspectos do desempenho e da segurança em um produto sem que interfiram, necessariamente, um no outro. Por exemplo, uma furadeira elétrica deve ser segura para seu usuário, particularmente em questões de isolamento elétrico, posição de botões de segurança e proteção contra emissão de cavacos ou partículas do objeto sendo perfurado. Por outro lado, e independentemente da segurança que a furadeira possa garantir ao operador, é interessante que ela também consiga ter um desempenho mínimo, ou seja, que ela consiga fazer um furo. Mas um aspecto não depende do outro. Diferentemente, após anos de discussão na IEC e em outros fóruns apropriados, chegou-se a um consenso que para equipamentos eletromédicos não seria possível dissociar o desempenho essencial da segurança básica que o equipamento deve garantir para ser considerado apropriado. Assim sendo, a filosofia da segurança e a filosofia do desempenho andam juntas na série de normas IEC 60601.
Todo equipamento eletromédico deve ser fabricado atentando para a segurança básica que ele deve prover para o usuário e para o paciente. A norma geral estabelece os requisitos mínimos gerais a serem atendidos pelos equipamentos em condições normais de uso ou uso equivocado, mas razoavelmente previsível (cláusula 4.1). Ou seja, o equipamento deve ser capaz de, mesmo sendo utilizado de maneira incorreta, manter-se seguro e com seu desempenho essencialmente inalterado. É de responsabilidade do fabricante, ainda, definir o que é desempenho essencial para seu equipamento eletromédico, podendo, neste caso, definir quais ensaios deverão ser aplicados no contexto da norma geral IEC 60601-1 (cláusula 4.3). Naturalmente, caso haja uma norma colateral ou particular que defina os ensaios para desempenho essencial, estas são prerrogativas inegáveis e não escamoteáveis, e não cabe ao fabricante optar por não atender aos seus requisitos técnicos. As partes do equipamento que entram em contato com o paciente devem ser consideradas potencialmente inseguras e devem ser ensaiadas adicionalmente ao corpo principal do equipamento, de acordo com o uso pretendido. Entretanto, também deve ser dada atenção às partes do equipamento que não estão previstas de entrar em contato com o paciente (4.6).
Todos os equipamentos devem ser construídos com gabinetes ou invólucros com resistência mecânica compatível com seu uso pretendido (4.9). O mesmo requisito se aplica aos fios elétricos (4.8) e demais partes, móveis ou não. Os ensaios mecânicos e elétricos, descritos no item 4, foram concebidos para avaliar a integridade, incluindo penetração de partículas de água, resistência à queda, rigidez dielétrica e corrente de fuga, entre outros. Como equipamentos eletromédicos dependem de energia elétrica para serem operados, quer sejam baterias ou rede elétrica diretamente, o fabricante deverá identificar quais ensaios deverão ser realizados para garantir que o suprimento de energia elétrica não comprometa a funcionalidade, segurança e usabilidade do seu produto (cláusulas 4.10 e 4.11).
A cláusula 4.7 da norma geral traz um conceito de suma importância na interpretação de como deve ser realizada a avaliação da segurança básica e do desempenho essencial dos equipamentos eletromédicos. Trata-se da “condição anormal sob uma só falha” (single fault condition) ou CASF. Todo equipamento eletromédico deve ser absolutamente livre de falhas, sem tolerâncias. Nenhuma falha pode ocorrer. Um liquidificador que não triture adequadamente uma banana para fazer uma vitamina pode até ser “perdoado”, já que a banana pode estar verde ou o usuário pode estar com pressa para preparar a vitamina, mas o mesmo não pode acontecer com um equipamento eletromédico. O fabricante, em sua análise de risco, deve prever todas as possibilidades em que o equipamento pode vir a falhar, ou seja, ser menos seguro do que o previsto ou não apresentar desempenho previsto, a fim de criar mecanismos para que essa falha não aconteça. Um equipamento eletromédico pode ser considerado livre de CASF se:
a)ele emprega meios efetivos para reduzir o risco de eventos adversos (por exemplo, isolamento elétrico duplo ou construção reforçada do seu gabinete);
b)ele for dotado de um meio de avisar o usuário caso uma falha ocorra a tempo de não prejudicar seu uso pretendido.
Para essa análise, o fabricante deverá adotar o gerenciamento de risco, incluindo a probabilidade e severidade de cada risco que possa causar CASF.
2.4. Gerenciamento de risco
Dentre todas as mudanças da terceira edição da IEC 60601-1, aquela de maior impacto é a implantação do gerenciamento de risco dos equipamentos eletromédicos por parte do fabricante. A norma a ser utilizada na elaboração do processo de gerenciamento de risco é a ISO 14971:2007 – Medical devices – Application of risk management to medical devices (82 páginas, elaborada pelo IEC/TC 210), com versão brasileira publicada pela ABNT como “ABNT NBR ISO 14971:2009 – Produtos para a saúde – Aplicação de gerenciamento de risco a produtos para a saúde” (88 páginas, elaborado pelo ABNT/CB-26).
Na norma geral da série 60601 há mais de 100 ocorrências de requisitos que fazem menção ao gerenciamento de risco. Essencialmente, o fabricante passa a ter uma certa liberdade de escolha em relação à forma como será feita a avaliação da conformidade do seu equipamento aos requisitos técnicos da norma geral. Sob a perspectiva dos preceitos do gerenciamento de risco segundo o que está normalizado, o fabricante poderá indicar quais ensaios são dispensáveis por não oferecerem risco acima de determinado limite aceitável. Caso o fabricante não seja capaz de evidenciar o gerenciamento de risco, o ensaio deverá ser feito rigorosa e completamente de acordo com os requisitos técnicos “tradicionais”. Ou seja, a avaliação do equipamento deve atender integralmente a todos os requisitos das normas geral, colaterais e particular aos quais está submetido.
A liberdade que essa abordagem traz para o fabricante não vem sem um custo adicional. Ao adotar o gerenciamento de risco para eliminar um determinado ensaio técnico, o fabricante deverá evidenciar competência completa do seu sistema da qualidade em gestão de risco seguindo estritamente o que está definido na ISO 14971. Ademais, o gerenciamento de risco deve ser feito em todo o ciclo de vida do produto, e não apenas na fase pré-mercado, ou seja, durante a concepção, o desenvolvimento e a fabricação do equipamento eletromédico. O fabricante deverá prover evidências de que o risco pós-mercado analisado, compreendendo as fases de marketing e venda, bem como o processo de vigilância pós-mercado, pode ocorrer durante a utilização e o descarte.
Um sistema de gerenciamento de risco inclui a identificação dos riscos em potencial e sua posterior análise do impacto na segurança e no desempenho, incluindo a avaliação e o controle. Cada etapa precisa ser apropriadamente identificada e avaliada, a fim de propor mecanismos de controle dos riscos identificados. Como um exemplo prático, vamos supor que um fabricante esteja desenvolvendo um equipamento de terapia por ultrassom (IEC 60601-2-5). O superaquecimento do cabeçote, parte do equipamento que transmite o ultrassom para o paciente, evidencia que há algum mau funcionamento. Esse mau funcionamento deve ser evitado por compreender um risco ao desempenho e à segurança do equipamento. A análise diz que, se superaquecer, o risco passa a ser o mau desempenho do equipamento ou a queimadura que pode ser provocada no paciente. O controle do risco deverá compreender, no mínimo, um circuito de proteção que impeça que o ultrassom continue a ser emitido, “cortando” o sinal elétrico que chega ao cabeçote. Mas o controle do risco poderá incluir, ainda, um registro da temperatura em função de outros parâmetros de ajuste do equipamento, o que permitiria, ex post facto, uma análise das condições de uso que causam o superaquecimento. Repare que, nesse exemplo, o gerenciamento de risco deveria ter sido realizado ainda na fase pré-mercado, durante o desenvolvimento do produto, mas sua aplicação compreende todo o ciclo de vida do produto, consciente de que o risco poderá aparecer durante a fase de mercado, isto é, durante a utilização pelo usuário final.
A abordagem de gerenciamento de risco se contrapõe, em tese, aos processos rígidos de ensaios com posterior análise passa/não passa (reprovação por não atender a critérios técnicos previstos nos requisitos normativos). Naturalmente, faz parte do gerenciamento de risco avaliar em quais requisitos técnicos o produto não pode deixar de ser avaliado. No caso do equipamento de terapia do exemplo anterior, a avaliação da área de radiação eficaz e a medição da potência ultrassônica (ver IEC 60601-2-5) continuarão imprescindíveis, e não há análise de risco que se sobreponha ao risco de ter uma emissão além ou aquém da prevista e necessária para o tratamento efetivo. Mas o aquecimento do cabeçote, como mencionado, é um risco adicional, concorrente aos demais requisitos de segurança básica e de desempenho essencial.
Toda análise de risco deve gerar um relatório, sendo parte integrante do gerenciamento de risco.
O item 4.2 da norma geral estabelece que os requisitos relativos ao gerenciamento de risco terão sido atendidos se o fabricante estabelecer um processo (ou procedimento) de gerenciamento de risco, se estabelecer critérios de níveis de risco aceitáveis e se demonstrar que eventuais riscos residuais são aceitáveis (considerando a política de risco aceitável que deve ser definida pelo fabricante no seu processo de gerenciamento de risco). Logo, está a cargo do fabricante estabelecer seu gerenciamento de risco, registrando de acordo com a norma internacional ISO 14791 (ou sua correspondente versão brasileira publicada pela ABNT).
O primeiro passo do gerenciamento de risco reside na definição do projeto do produto (equipamento eletromédico). Devem ser considerados o uso pretendido, o ambiente de uso, as características do usuário, o paciente que será atendido pelo equipamento e demais características que irão definir o modelo de desenvolvimento tecnológico do produto. Para cada uma dessas perspectivas, o fabricante irá identificar potenciais riscos, ou seja, situações perigosas que poderão ocasionar desvios de funcionamento do equipamento. Depois, seguem-se as subsequentes etapas de gerenciamento de risco. Importante mencionar que, como alertado anteriormente, o gerenciamento de risco perdura por todo o ciclo de vida do produto. Portanto, cabe ao fabricante criar mecanismos de acompanhamento do produto no mercado, identificando oportunidades de melhoria e implantando-as, inclusive no projeto de desenvolvimento.
A seguir são apresentados os principais termos e definições da ABNT NBR ISO 14971:2009 relacionados ao gerenciamento de risco. A numeração que precede o termo definido é a respectiva cláusula na norma em questão, e o termo em parênteses é o termo original em inglês.
✓2.2 Dano (harm): lesão física ou prejuízo à saúde da pessoa, ou prejuízo à propriedade ou ao meio ambiente (ISO/IEC Guide 51:1999, definição 3.3).
✓2.3 Perigo (hazard): fonte potencial de dano (ISO/IEC Guide 51:1999, definição 3.5).
✓2.4 Situação perigosa (hazardous situation): circunstância em que pessoa, propriedade e meio ambiente estejam expostos a um ou mais perigos (ISO/IEC Guide 51:1999, definição 3.6).
✓2.5 Utilização destinada/Propósito destinado (intended use/intended purpose): utilização para qual um produto, processo ou serviço é destinado de acordo com suas especificações, instruções e informações oferecidas pelo fabricante.
✓2.7 Ciclo de vida (life-cycle): todas as fases de vida de um produto para saúde desde a concepção inicial até a retirada de serviço e descarte.
✓2.8 Fabricante (manufacturer): pessoa física ou jurídica responsável pelo projeto, pela fabricação, pela embalagem ou pela rotulagem de um produto para a saúde, montagem de um sistema ou adaptação de um produto antes de ser colocado no mercado ou em funcionamento, independentemente do fato dessas operações serem realizadas por essa pessoa ou em seu nome, por uma terceira parte.
✓2.9 Produto para a saúde (medical device): qualquer instrumento, aparelho, implemento, máquina, produto, implante ou calibrador in vitro, software, material ou outro artigo similar ou relacionado pelo fabricante a ser utilizado, sozinho ou em combinação, em seres humanos, para um ou mais propósitos específicos (ver ABNT NBR ISO 14971:2009, item 2.9).
✓2.10 Evidências objetivas (objective evidence): dados que apoiam a existência ou veracidade de alguma coisa.
✓2.11 Pós-produção (post-production): parte do ciclo de vida do produto após o projeto ter sido completado e o produto ter sido fabricado.
✓2.15 Risco residual (residual risk): risco remanescente após as medidas de controle de risco terem sido adotadas (ISO/IEC Guide 51:1999, definição 3.9).
✓2.16 Risco (risk): combinação da probabilidade de ocorrência de um dano e a severidade de tal dano (ISO/IEC Guide 51:1999, definição 3.2).
✓2.17 Análise de risco (risk analysis): utilização sistemática de informação disponível para identificar perigos e estimar riscos (ISO/IEC Guide 51:1999, definição 3.10).
✓2.18 Determinação de risco (risk assessment): processo completo composto pela análise e avaliação de risco (ISO/IEC Guide 51:1999, definição 3.12).
✓2.19 Controle de risco (risk control): processo por meio do qual decisões são tomadas e medidas são implementadas para a redução ou manutenção de riscos dentro de níveis especificados.
✓2.20 Estimativa de risco (risk estimation): processo utilizado para designar valores à probabilidade de ocorrência do dano e à sua severidade.
✓2.21 Avaliação de risco (risk evaluation): processo de análise de relação entre o risco estimado e dado critério de risco para determinar a sua aceitabilidade.
✓2.22 Gerenciamento de risco (risk management): aplicação sistemática de políticas e práticas de gerenciamento às tarefas de análise, avaliação, controle e monitoração de risco.
✓2.23 Arquivo de gerenciamento de risco (risk management file): conjunto de registros e outros documentos que são produzidos pelo gerenciamento de risco.
✓2.24 Segurança (safety): ausência de riscos inaceitáveis (ISO/IEC Guide 51:1999, definição 3.1).
✓2.25 Severidade (severity): medida das possíveis consequências de um perigo.
A Figura 4 apresenta um modelo de matriz de risco. A ponderação entre as categorias apresentadas nos eixos horizontal e vertical faz parte da análise de risco que deve ser realizada dentro do processo de gerenciamento de risco. Portanto, as regiões de riscos aceitáveis, que demandam medidas de redução ou que não são aceitáveis, podem variar para cada risco em consideração.
As condições anormais sob uma só falha (CASF) são analisadas com base no modelo de matriz de risco. Cada tipo de CASF é analisada conforme seu comprometimento com a segurança básica e o desempenho essencial, sendo estas as premissas fundamentais da norma geral IEC 60601-1.
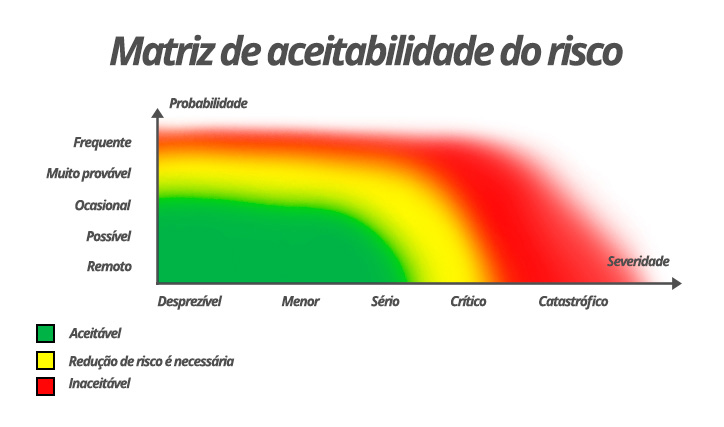
Figura 4. Modelo de matriz de risco de um produto. Fonte: elaborada pela Sociedade Brasileira de Metrologia (SBM). Direitos autorais cedidos para esta obra.
A posição em que o risco deve estar é na região aceitável. A região na qual foram identificados riscos, mas que não são inaceitáveis, determina quais ações de redução de risco devem ser realizadas. Em situações muito particulares, devidamente justificadas e embasadas tecnicamente, pode-se definir uma situação denominada ALARP, do acrônimo em inglês As Low As Reasonably Practicable, ou seja, “tão baixo quanto razoavelmente praticável”.
Como identificar os perigos ou possíveis fontes de falha? Há extensa literatura a respeito, mas, em linhas gerais, as seguintes fontes de identificação de riscos de falha podem ser elencadas: usuários, médicos ou agentes de saúde, literatura científica, alertas de segurança, normas de segurança e desempenho (incluindo as da série 60601), informações públicas de produtos concorrentes, experiência de campo de produtos similares, opiniões de especialistas, efeitos clínicos nos modos de falha e legislação.
2.5. Ciclo de vida
Ciclo de vida é um conceito aplicado no desenvolvimento do modelo de gerenciamento de risco dos equipamentos eletromédicos. A Figura 5 apresenta um modelo esquemático do que deve ser analisado no ciclo de vida.
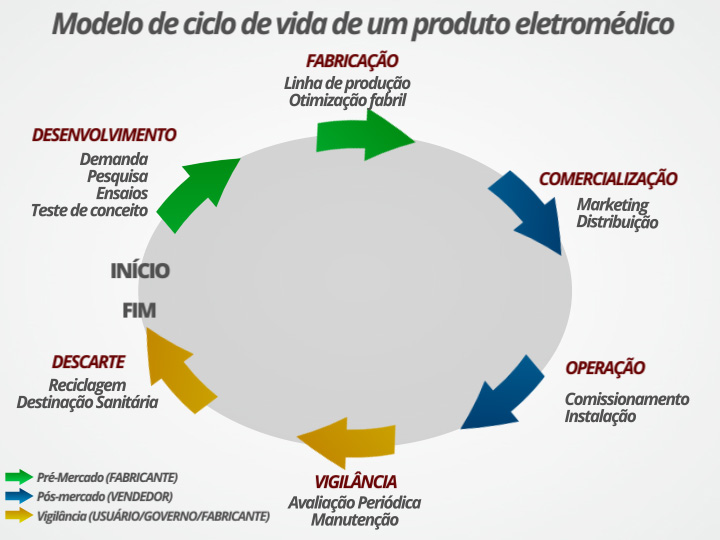
Figura 5. Modelo de ciclo de vida de um produto eletromédico. Fonte: elaborada pela Sociedade Brasileira de Metrologia (SBM). Direitos autorais cedidos para esta obra.
O início da vida de um equipamento eletromédico começa, como praticamente todo produto, com a observação de uma demanda da sociedade ou, especificamente, do setor de saúde. Nessa fase de concepção e de desenvolvimento, devem ser capturadas as necessidades dos clientes e devem ser observados os requisitos legais e técnicos de projeto. A empresa (fabricante) deve se estruturar, formar recursos humanos, desenvolver o produto ou identificar fornecedores e parceiros, entre outras ações. O desenvolvimento vem logo a seguir, etapa na qual são realizados os ensaios preliminares, os testes de conceito, o desenvolvimento de protótipos e a preparação da documentação preliminar do produto. Geralmente, as instâncias competentes para autorizar a comercialização, tais como Anvisa e Inmetro, deverão ser acionadas também nessa etapa de desenvolvimento. Caso contrário, o projeto poderá ser inviabilizado por alguma questão formal regulatória ou de avaliação da conformidade. A certificação do equipamento segundo o esquema de avaliação da conformidade do Inmetro e a obtenção do registro junto à Anvisa acontecem nessa etapa. O final dessa etapa do ciclo de vida, aquela que depende praticamente apenas do fabricante, se encerra com a fabricação propriamente dita. Essa fase é denominada “pré-mercado”.
Na sequência, o ciclo de vida entra na fase “pós-mercado”. Os esforços se concentram na comercialização, o que inclui, mas não se limita a, ações de marketing, distribuição e armazenamento do produto. O cliente passa a ser buscado não mais apenas para definir as especificações do produto, como acontece no desenvolvimento, mas para efetivamente comprar. O tratamento deve ser prospectivo, mas não necessariamente agressivo, considerando-se a concorrência. Imediatamente após a compra, muitas vezes se mesclando quanto ao momento em que ocorre e sobre quem tem a responsabilidade de realizar a atividade, acontece a entrada em operação do equipamento. O comissionamento, que inclui a instalação, eventualmente com adequação de espaço físico, é vital para a boa operação de qualquer equipamento eletromédico. O treinamento também pode ser parte integrante dessa etapa e poderá fazer toda a diferença entre uma boa e uma má operação.
A vigilância ocorre desde o momento em que o equipamento é colocado em operação. Sua avaliação continuada e constante deve assegurar o bom funcionamento do equipamento, livrando-o de falhas previstas no projeto. A vigilância pode ter caráter formal regulatório, no caso de estar prevista nos requisitos de avaliação da conformidade ou em regulamentos técnicos que regem sua certificação, ou de análise do ambiente no qual o equipamento está instalado. As manutenções, tanto periódicas (preventivas) como esporádicas (corretivas), devem ser seguidas de uma avaliação sobre as condições de funcionamento do equipamento. É fundamental garantir que os requisitos de segurança básica e de desempenho essencial sejam mantidos após as intervenções. Normalmente, deverão ser realizados ensaios por especialistas, preferencialmente por laboratórios acreditados e por técnicos competentes.
O descarte pode ser uma etapa extremamente crítica do ciclo de vida. Por exemplo, equipamentos que utilizam fontes de radiação nuclear devem ter um tratamento diferenciado na etapa do descarte. Todos se lembram do desastre ocorrido em Goiânia quando fontes de Césio-137 foram encontradas em lixo comum (<https://pt.wikipedia.org/wiki/Acidente_radiológico_de_Goiânia>). Mas não são apenas fontes radioativas que devem ser descartadas com cuidado diferenciado. Baterias, incluindo as de chumbo-ácido ou de níquel-cádmio, são igualmente nocivas ao meio ambiente e à saúde das pessoas. Seus riscos devem ser rigorosamente analisados no gerenciamento de risco. A possiblidade de reciclagem ou de reutilização da matéria-prima e de componentes dos equipamentos eletromédicos deve ser igualmente avaliada a fim de minimizar os impactos ambiental e econômico do equipamento após seu descarte.
O gerenciamento de risco, se bem feito de acordo com os preceitos das normas técnicas IEC 60601-1 e ISO 14791, deve considerar cada fase, etapa e subetapa do ciclo de vida do equipamento eletromédico.
2.6. Classificação dos equipamentos médicos
A cláusula 6 da norma IEC 60601-1 descreve os diversos aspectos segundo os quais os equipamentos eletromédicos devem ser classificados. Essa classificação deve incluir todas partes e acessórios que compõem o equipamento ou sistema.
A cláusula 6.2 classifica o equipamento em relação ao risco de choques elétricos. Os equipamentos que são alimentados diretamente da rede elétrica podem ser Classe I ou Classe II. Os equipamentos alimentados de maneira autônoma por bateria são classificados como “energizado internamente”, embora, no caso de precisarem ser recarregados conectando-se a fonte externa de energia elétrica, devam ser classificados como Classe I ou Classe II enquanto estiverem conectados. Equipamentos do tipo Classe I apresentam isolamento ou aterramento adicional para todas as partes metálicas ou com acesso facilitado (janelas de inspeção, por exemplo), além do isolamento normal das partes elétricas. Todos os componentes, partes ou acessórios passíveis de carregamento elétrico são ligados ao aterramento do local de uso. Os equipamentos Classe II, além da proteção dos equipamentos Classe I, dispõem de precauções adicionais contra choque elétrico, tais como isolamento duplo ou reforçado.
Segundo a cláusula 6.3, os equipamentos eletromédicos são classificados quanto ao ingresso de água e de partículas potencialmente danosas. A classificação é feita segundo a norma técnica IEC 60529 – Degrees of protection provided by enclosures (IP Code), 207 páginas, atualmente na edição 2.2, publicada em 2013 e elaborada pelo IEC/TC70, sendo no Brasil a ABNT NBR IEC 60529:2005 Errata 2:2011 – Graus de proteção para invólucros de equipamentos elétricos (código IP). O “código IP” assume as formas IPN1N2, onde N1 é o grau de proteção contra partículas materiais (ou X), e N2 é o grau de proteção contra água (ou X). O “X” significa que o equipamento não foi avaliado quanto à proteção para o quesito em questão. Por exemplo, se um equipamento é classificado como IPX7, ele tem proteção grau 7 para penetração de água e não foi avaliado para penetração de outros agentes (partículas sólidas).
Quadro 3. Classificação IP e seu significado.
|
Resistência a objetos sólidos e poeira |
Resistência à água |
||
|
IP0X |
Nenhuma proteção especial |
IPX0 |
Nenhuma proteção especial |
|
IP1X |
Proteção contra objetos sólidos > 50 mm de diâmetro |
IPX1 |
Proteção contra gotas de água |
|
IP2X |
Proteção contra objetos sólidos > 12,5 mm de diâmetro |
IPX2 |
Proteção contra gotas de água quando estiver inclinado a até 15 graus em relação à posição normal |
|
IP3X |
Proteção contra objetos sólidos > 2,5 mm de diâmetro |
IPX3 |
Proteção contra borrifos de água |
|
IP4X |
Proteção contra objetos sólidos > 1 mm de diâmetro |
IPX4 |
Proteção contra respingos de água |
|
IP5X |
Proteção contra poeira; entrada limitada (nenhum depósito prejudicial) |
IPX5 |
Proteção contra jatos d’água de baixa pressão durante três minutos, no mínimo |
|
IP6X |
À prova de poeira |
IPX6 |
Proteção contra jatos fortes durante três minutos, no mínimo |
|
IPX7 |
Proteção contra os efeitos da imersão até 1 metro de água durante trinta minutos |
||
|
IPX8 |
Proteção contra os efeitos da imersão contínua em água a uma profundidade de mais de 1 metro. As condições exatas são especificadas pelo fabricante de cada dispositivo |
||
Fonte: elaboração própria.
Segundo a cláusula 6.4, os equipamentos eletromédicos devem ser classificados de acordo com a forma como a esterilização de suas partes e componentes deve ser feita. Como exemplo, o equipamento pode ser esterilizado por gás de óxido de etileno, por irradiação tipo raio gama, por calor úmido em autoclave ou por outros métodos validados e descritos pelo fabricante.
Caso o equipamento possa ser utilizado em atmosfera rica em oxigênio (potencialmente mais explosiva), ele deve ser classificado e identificado como tal (cláusula 6.5). Por fim, o equipamento deve ser classificado como de operação contínua ou intermitente (não contínua), conforme requisito da cláusula 6.6.
2.7. Identificação, marcação e documentação
Os equipamentos eletromédicos devem apresentar identificação, marcas e documentação apropriadas, de acordo com sua classificação e funcionalidade, além de alertas de perigo ou de modo de operação, conforme a determinação de sua análise de risco. As normas colaterais e particulares podem trazer informações adicionais quanto às marcações e identificações. Porém, em relação à norma geral, as marcas devem estar presentes na parte externa do equipamento (cláusula 7.2), interna (cláusula 7.3) ou nos acessórios, controles e partes complementares (cláusula 7.4).
Em linhas gerais, todas as marcas, sinais e avisos devem ser claramente visíveis. Para tanto, o operador deve ser capaz de ver e identificar a marca a, pelo menos, 1 metro de distância e em um ângulo de até 30° em relação à direção perpendicular à superfície onde a marca se encontra. Marcas e sinais devem ser duráveis por toda a vida útil prevista para o equipamento. Para avaliar a durabilidade, a marca deve ser esfregada gentilmente com um pano embebido com água destilada por 15 segundos e, depois de seca, com um pano embebido com metilato também por 15 segundos e, por fim, por mais 15 segundos com álcool isopropílico.
Todos os equipamentos eletromédicos devem conter a marca e o modelo em seu exterior, salvos os casos em que a omissão dessa informação não cause risco (veja matriz de riscos na Figura 4). Nos casos em que a marcação não seja possível (por exemplo, por ser o gabinete demasiadamente pequeno), essas informações devem ficar claras na documentação acompanhante. Os acessórios devem ser igualmente identificados.
Caso o equipamento eletromédico seja energizado por outro equipamento que a ele se conecte, deve ser afixada uma marca no local mais viavelmente próximo do ponto de conexão entre os equipamentos. Naqueles equipamentos alimentados diretamente da rede elétrica, deve ser afixada uma etiqueta próxima ao ponto de conexão com a eletricidade, no gabinete ou onde mais facilmente essa informação possa ser identificada, a fim de informar os limites de tensão elétrica. Para equipamentos que fiquem instalados definitivamente em determinado local, ou seja, não sujeitos a eventual desconexão da rede elétrica por parte do usuário, essa marca pode ficar internamente afixada. Os pontos funcionais de aterramento e os terminais de alta tensão devem ser apropriadamente identificados.
A marcação IP, conforme sua classificação, deve ficar visível, exceto para os equipamentos classificados como IPX0 (sem qualquer isolamento ou proteção contra água) ou IP0X (sem proteção contra partículas sólidas). Essa observação é particularmente importante. Caso a etiqueta informando a classificação IP do equipamento não esteja visível, ou tenha sido inadvertidamente retirada, ou ainda tenha estragado com o uso, o usuário poderá achar que o equipamento não possui qualquer capacidade de evitar penetração de água ou partículas em seu interior.
Uma marcação informando se o equipamento pode ou não ser operado ininterruptamente deve estar presente. Na ausência dessa marcação, o equipamento deve ser considerado apropriado para uso ininterrupto, o que pode vir a ser um problema caso a etiqueta tenha sido danificada ou retirada inapropriadamente.
Os equipamentos que possam vir a gerar efeitos fisiológicos adversos nos pacientes ou usuários, mas cuja operação típica não leve a essa constatação naturalmente, também deverão conter marcações apropriadas. O tipo e efeito fisiológico devem ser informados, utilizando-se a simbologia normalizada.
Outras marcações externas incluem o tipo de refrigeração ao qual o equipamento deve estar submetido, o tipo de embalagem protetora (para transporte e armazenamento), as fontes externas de pressão (ar comprimido, por exemplo) e estabilidade mecânica. Essas marcações externas podem conter também outros tipos de informações.
As marcações internas devem estar claramente visíveis quando o fechamento do gabinete for removido. As marcações e as etiquetas devem incluir pontos quentes, ou seja, aqueles pontos cuja temperatura superficial possa causar danos ao usuário. Tal qual acontece nas marcações externas, os pontos de alta tensão devem ser claramente identificáveis no interior dos equipamentos, assim como as baterias, localização dos fusíveis elétricos e os pontos de fuga de corrente de segurança.

Figura 6. Exemplo de sinalização de perigo para o usuário. Fonte: elaborada pela Sociedade Brasileira de Metrologia (SBM). Direitos autorais cedidos para esta obra.
Todos os controles e instrumentos devem ser marcados apropriadamente quanto aos seus interruptores (botões de ligar e desligar) e posições de ajuste. Preferencialmente, devem ser utilizadas as unidades do Sistema Internacional de Unidades (SI) para os controles dos equipamentos eletromédicos. Todas as chances de uso indevido ou de ajuste que possam vir a causar danos devem ser evitadas, sendo esses riscos devidamente informados nas marcas dos controles.
As cláusulas 7.5 e 7.6 apresentam os sinais e símbolos de segurança obrigatórios. Os símbolos devem seguir o previsto na norma técnica ISO 7010 – Graphical symbols – Safety colours and safety signs – Registered safety signs (símbolos gráficos – cores de segurança e sinais de segurança – sinais de segurança registrados).

Figura 7. Exemplo de símbolos da norma técnica ISO 7010. Fonte: elaborada pela Sociedade Brasileira de Metrologia (SBM). Direitos autorais cedidos para esta obra.
A cláusula 7.7 estabelece as cores para os fios elétricos a serem utilizados nos equipamentos eletromédicos. Todos os condutores de proteção de aterramento devem ser revestidos por material isolante nas cores verde e amarelo ao longo de todo o seu comprimento. O terminal elétrico neutro deve ser na cor azul clara, enquanto os fios de energia devem seguir o regramento previsto nas normas IEC 60227-1 – Polyvinyl chloride insulated cables of rated voltages up to and including 450/750 V – Part 1: General requirements (Cabos isolados com cloreto de polivinil com tensões nominais até 450/750 V inclusive – Parte 1: Requisitos gerais) ou IEC 60245-1 – Rubber insulated cables – Rated voltages up to and including 450/750 V – Part 1: General requirements (Cabos isolados de borracha – Tensões nominais até 450/750 V inclusive – Parte 1: Requisitos gerais).
A cláusula 7.8 exemplifica as cores que os avisos e alarmes de segurança devem ter. A cor vermelha indica que o operador deve tomar alguma ação imediata sob o risco de falha ou de danos. A cor amarela indica que o operador deve ser ágil, mas não necessita ação imediata como demanda o alerta vermelho. A cor verde indica que o sistema ou atividade estão prontos para serem realizados e estão em condições normais de operação. Outras cores podem ser utilizadas, desde que tenham significado distinto dos sinais vermelho, amarelo ou verde, e devem ser detalhadamente explicadas quanto à sua funcionalidade.

Figura 8. Identificação e marcação devem estar claras e visíveis para o usuário. Fonte: elaborada pela Sociedade Brasileira de Metrologia (SBM). Direitos autorais cedidos para esta obra.
Além de sinais e marcações relevantes para evitar riscos operacionais, os equipamentos eletromédicos devem ser dotados de documentos acompanhantes completos. A documentação deve conter, pelo menos, informações sobre o uso do equipamento, os avisos e alertas de segurança, a forma de alimentação elétrica, os cuidados particulares com essa alimentação, a descrição geral e detalhada do equipamento, a forma de instalação e de operação, o procedimento para iniciar (ligar) e finalizar a operação (desligar), o modo de realizar limpeza e a desinfecção. Também se incluem cuidados adicionais como manutenção, proteção ambiental e descrição técnica detalhada.
2.8. Inspeção visual
A inspeção visual é uma abordagem utilizada em diversas áreas técnicas, inclusive Ensaios Não Destrutivos. No contexto da norma geral, a inspeção visual é mencionada como sendo o procedimento para se avaliar o atendimento a requisitos. Dentre eles, elencam-se os seguintes:
✓Gabinete ou recipiente que protege as partes internas do equipamento. A inspeção visual deve ser realizada a fim de avaliar eventuais danos ou avarias, tais como rachaduras, duros, amassados ou mossas, por exemplo.
✓Obstrução mecânica. Deve ser feita a atenta avaliação das boas condições e a livre movimentação das partes móveis, conectores, soquetes, botões de acionamento, teclas avulsas e teclados. As partes mecânicas devem ser inspecionadas quanto a sua integridade com a mesma atenção que o gabinete.
✓Cabeamento. Os cabos de alimentação elétrica devem ser inspecionados a fim de se garantir que não há fios desencapados, cortados ou quebrados. Também devem ser inspecionados os cabos que conectam partes móveis ou remotas, tais como transdutores ou acessórios.
✓Marcas e etiquetas. Elas devem ser atentamente inspecionadas, conferindo sua conformidade com as informações apresentadas nos manuais e demais documentos acompanhantes. As marcas e etiquetas são fundamentais por comunicarem visualmente tanto o equipamento quanto suas partes específicas.
2.9. Finalizando
Neste capítulo foram apresentados os principais conceitos e a filosofia da norma geral da série IEC 60601. A norma geral é complementada pelas oito normas colaterais, que, juntas, formam o conjunto de requisitos para a segurança básica e o desempenho essencial para equipamentos eletromédicos. No próximo capítulo, todas as normas colaterais serão apresentadas.
2.10. Para fixar o conteúdo
A fim de promover um melhor entendimento sobre a norma geral, convide colegas de estudo para pesquisar nas fontes apresentadas na bibliografia no final do livro ou estude sozinho e discuta as questões apresentadas a seguir.
Exercício 4. Conceitualmente, o que há de novo na terceira edição da norma geral em relação às edições anteriores? Como essa nova filosofia impacta no desenvolvimento de novos equipamentos eletromédicos?
Exercício 5. Qual a importância do ciclo de vida do equipamento eletromédico? Descreva a responsabilidade de cada ator nas diversas etapas do ciclo de vida de um equipamento eletromédico.
Exercício 6. Escolha um equipamento eletromédico com o qual você tenha familiaridade. Classifique-o de acordo com a cláusula 6 da norma geral da série IEC 60601.
Exercício 7. Observe um equipamento eletromédico que esteja em uso. Veja se todas as identificações e marcações previstas na cláusula 7 da norma geral estão presentes.